Abstract
p = 3.97e-01 (Mann-Whitney U)
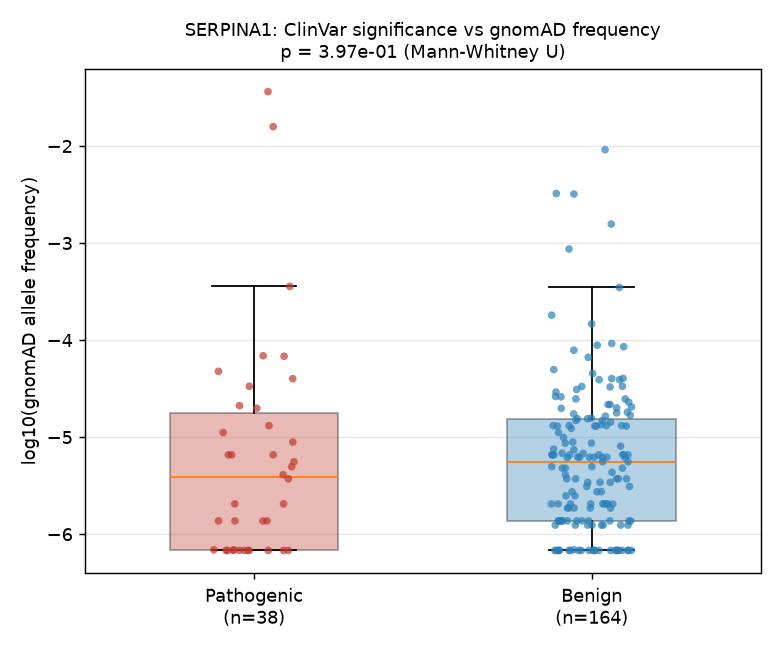
We tested whether variants classified as pathogenic or likely pathogenic by ClinVar for the SERPINA1 gene have lower population allele frequencies in gnomAD than ClinVar-classified benign or likely benign variants. After merging 316 classified ClinVar records with 1,616 gnomAD variants via genomic locus (chrom:pos), 202 variants overlapped (38 pathogenic, 164 benign). A two-sided Mann-Whitney U test on allele frequencies yielded U = 2841.5, p = 0.397, which is not statistically significant at the 0.05 threshold. While the median allele frequency of pathogenic variants (3.91 × 10⁻⁶) was numerically lower than that of benign variants (5.58 × 10⁻⁶), this difference did not reach statistical significance. Our results do not provide evidence that ClinVar pathogenic variants in SERPINA1 are systematically rarer in the general population than benign variants, though the small sample size limits statistical power.
[withheld: unsafe public value]
not recorded — legacy attempt
722559d586880cac990f8a5552986b941d6a36459b17297e290a1bff24a3a12d
edcf67e94c93ee9002c7a762caa46984c9aff7c1
{"gene":"SERPINA1","seed":1234,"reference_template":"clinvar_gnomad_ensembl","join_key":"genomic locus chrom:pos (ref/alt-agnostic)","n_clinvar_classified":316,"n_merged_with_gnomad_af":202,"n_pathogenic":38,"n_benign":164,"outcome":"success","test":"Mann-Whitney U (two-sided) on allele frequency","u_statistic":2841.5,"p_value":0.39748591672845146,"median_af_pathogenic":0.00000391,"median_af_benign":0.00000558,"direction":"pathogenic rarer","significant_at_0.05":false,"headline_statistic":"p = 3.97e-01 (Mann-Whitney U)"}