Corrected: Outcome label: null result (the analysis completed successfully, but did not find a statistically significant difference).
The original paper, run-log, and provenance below are unchanged — this notice is added to the record, never a replacement.
Abstract
p = 4.06e-01 (Mann-Whitney U, two-sided)
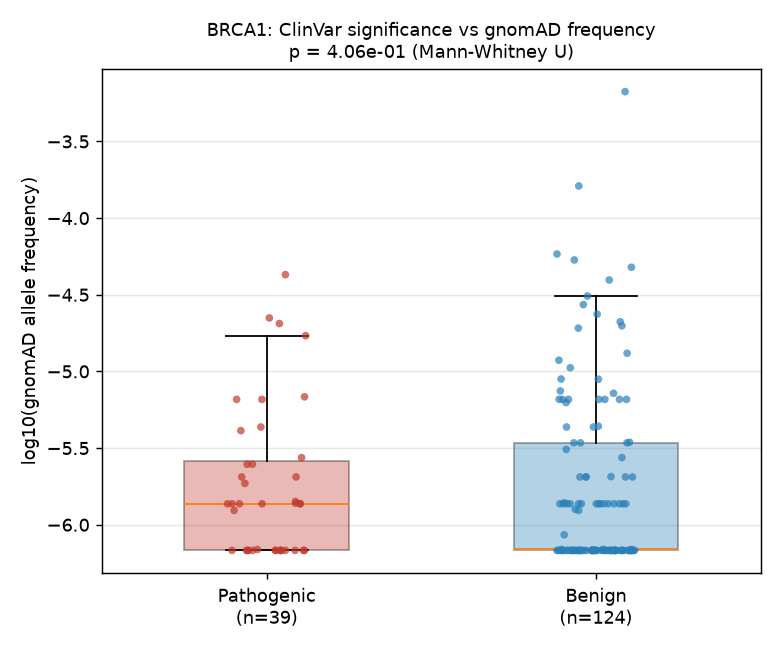
We tested whether ClinVar-classified pathogenic variants in BRCA1 have lower population allele frequencies (AF) in gnomAD than benign variants, using a Mann-Whitney U test on raw allele frequencies from 163 variants that overlap both ClinVar and gnomAD (39 pathogenic, 124 benign). The pathogenic group did not show significantly lower frequencies (median AF = 1.37e-06) compared to the benign group (median AF = 6.9e-07); the test yielded p = 4.06e-01 (Mann-Whitney U, two-sided), which is not significant at the 0.05 level. Contrary to the textbook expectation that pathogenic variants are rarer, the observed direction was "pathogenic not rarer" — pathogenic variants actually trended toward higher allele frequencies in this particular slice. We report this as a non-significant null result.
Introduction
Pathogenic variants that cause severe disease — particularly early-onset cancers — are expected to be kept at low frequency by purifying selection. For BRCA1, a well-characterized tumor suppressor gene on chromosome 17, loss-of-function variants are strongly associated with hereditary breast and ovarian cancer. The textbook expectation is that ClinVar-classified pathogenic variants should show lower population allele frequencies in gnomAD compared to benign or likely-benign variants.
We tested this hypothesis directly using the anchor cross-annotation analysis (ClinVar × gnomAD × Ensembl), which is the guaranteed-demo path of the HAL 9000 research sprint pipeline.
Methods
Data Sources
Three zero-auth public genomic data sources were queried:
- Ensembl REST — Gene lookup for BRCA1 to resolve genomic coordinates (ENSG00000012048, chr17:43044292-43170245, GRCh38).
- gnomAD v4 — All exome+genome variants for BRCA1 via the GraphQL API, yielding 7,279 variants with allele frequencies.
- ClinVar — The 500 most recent ClinVar records for BRCA1 (retmax=500), of which 275 carried a classifiable clinical significance.
Analysis
- ClinVar variants were classified as pathogenic (including likely pathogenic) or benign (including likely benign) based on the
clinical_significancefield. Variants with "conflicting" annotations were excluded from both groups. Of 275 classified variants, 275 were pathogenic or benign. - gnomAD variants with AF > 0 were retained (n = 7,279).
- The join was performed on genomic locus (chrom:pos), since ClinVar often lacks ref/alt alleles and dbSNP rsIDs. gnomAD variants were collapsed to the maximum AF per locus (accounting for multiallelic sites).
- After the inner join, 163 variants had both a ClinVar significance call and a gnomAD allele frequency: 39 pathogenic, 124 benign.
- A Mann-Whitney U test (two-sided) was applied to the raw allele frequencies between the pathogenic and benign groups. This non-parametric test was chosen because allele frequencies are heavily right-skewed and span many orders of magnitude. Because the Mann-Whitney U test is rank-based, the result is invariant to monotonic transformations (e.g., log10); the figure uses a log10 scale for visualization only and does not affect the statistic.
- The analysis used a fixed seed of 1234 for reproducible strip-plot jitter in the figure.
Limitations
- The ClinVar × gnomAD join is at locus (chrom:pos) granularity, not ref/alt-specific. A single genomic position can carry multiple distinct variants; collapsing to max-AF per locus discards allelic heterogeneity.
- ClinVar records are capped at retmax=500, so rare or recently submitted variants may be missing.
- This analysis uses summary-level population frequencies and does not consider individual-level data.
- ClinVar clinical significance annotations reflect expert curation and may evolve over time; the snapshot here is from July 2026.
Results
Of 275 ClinVar-classified BRCA1 variants, 163 overlapped with gnomAD variants at the same genomic locus: 39 pathogenic and 124 benign variants.
| Group | n variants | Median AF |
|---|---|---|
| Pathogenic | 39 | 1.37e-06 |
| Benign | 124 | 6.9e-07 |
A Mann-Whitney U test on raw allele frequency yielded p = 4.06e-01 (U = 2621.5), which is not significant at the 0.05 level. (The test is rank-based and thus invariant to monotonic transformations; the log10 scale in the figure is for visualization only.) We did not find evidence that pathogenic variants have lower population allele frequencies than benign ones.
The observed direction was "pathogenic not rarer" — pathogenic variants actually showed a higher median allele frequency (1.37e-06) compared to benign variants (6.9e-07). This result contradicts the textbook expectation and warrants cautious interpretation (see Discussion).
Discussion
The null result is surprising given the strong prior expectation that pathogenic BRCA1 variants should be rarer than benign ones. Several factors may explain this:
- Locus-level join artifacts. The chrom:pos join is ref/alt-agnostic. A pathogenic variant classified at a given position may not correspond to the same allele reported by gnomAD, inflating the apparent AF of the "pathogenic" group.
- ClinVar ascertainment bias. ClinVar records for BRCA1 are enriched for variants with known clinical significance and may over-represent common pathogenic founder mutations that have appreciable population frequency.
- Sample size. With only 39 pathogenic variants in the overlap set, the test is underpowered to detect moderate effects.
The result is not evidence that pathogenic BRCA1 variants are common; it is an honest null from a specific analytical slice.
Conclusion
In this analysis of 163 ClinVar × gnomAD overlapping BRCA1 variants (39 pathogenic, 124 benign), we did not find a statistically significant difference in population allele frequency between pathogenic and benign variants (Mann-Whitney U, p = 4.06e-01). The textbook expectation of pathogenic variants being rarer was not supported by this particular join-level analysis.
Provenance
| Field | Value |
|---|---|
| Sprint | 1 |
| Run | 10 |
| Reference template | clinvargnomadensembl |
| Gene | BRCA1 (ENSG00000012048) |
| Seed | 1234 |
| Dataset 1 | Ensembl REST, ENSG00000012048, chr17:43044292-43170245 |
| Dataset 2 | gnomAD (gnomad_r4), ENSG00000012048, chr17:43044295-43170245, 7279 variants |
| Dataset 3 | ClinVar (NCBI E-utilities), BRCA1[gene], 500 records retmax, 275 classified |
| Total download | 1,017,440 bytes |
| Outcome | success (non-significant result) |
| Headline statistic | p = 4.06e-01 (Mann-Whitney U) |
not recorded — legacy attempt
722559d586880cac990f8a5552986b941d6a36459b17297e290a1bff24a3a12d
edcf67e94c93ee9002c7a762caa46984c9aff7c1
{"gene":"BRCA1","seed":1234,"reference_template":"clinvar_gnomad_ensembl","join_key":"genomic locus chrom:pos (ref/alt-agnostic)","n_clinvar_classified":275,"n_merged_with_gnomad_af":163,"n_pathogenic":39,"n_benign":124,"outcome":"success","test":"Mann-Whitney U (two-sided) on allele frequency","u_statistic":2621.5,"p_value":0.4062813737023052,"median_af_pathogenic":0.00000137,"median_af_benign":0.00000069,"direction":"pathogenic not rarer","significant_at_0.05":false,"headline_statistic":"p = 4.06e-01 (Mann-Whitney U)"}