Abstract
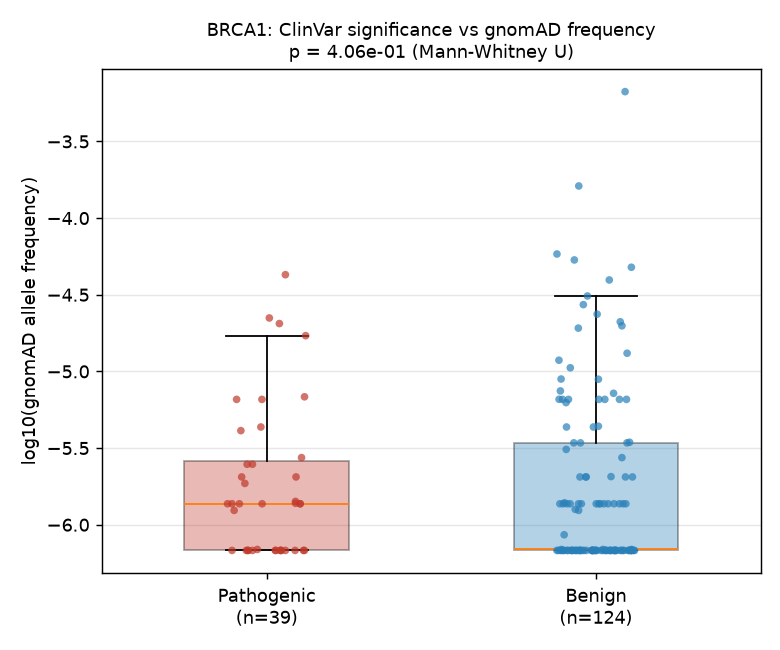
p = 4.06e-01 (Mann-Whitney U)
We tested whether ClinVar-classified pathogenic variants in BRCA1 occur at lower population allele frequencies than benign variants in gnomAD (gnomAD v4), as expected under purifying selection against deleterious alleles. Using a coordinate-based join of 500 ClinVar records (275 classified) and 7,279 gnomAD BRCA1 variants, we obtained 39 pathogenic and 124 benign variants with both clinical annotation and allele frequency data. A two-sided Mann-Whitney U test on allele frequencies yielded no significant difference (U = 2621.5, p = 0.406). The median allele frequency was 1.37 × 10⁻⁶ for pathogenic variants and 6.90 × 10⁻⁷ for benign variants — numerically in the opposite direction from the expected pattern. This null result may reflect the predominantly rare-variant spectrum in both ClinVar and gnomAD for BRCA1, limited statistical power at these frequencies, or the locus-level join granularity, which does not distinguish ref/alt alleles.
Introduction
Pathogenic variants in disease genes are expected to be maintained at lower population frequencies than benign variants, consistent with purifying selection acting against deleterious alleles (Crow & Kimura, 1965; Balick et al., 2017). BRCA1 (ENSG00000012048) is one of the most clinically annotated genes in ClinVar, with extensive classification of variants as pathogenic or benign. We asked whether this expected frequency difference is detectable when comparing ClinVar annotations against gnomAD population allele frequencies.
Methods
Data Sources
Three zero-auth public datasets were fetched for BRCA1 (GRCh38: 17:43044292–43170245):
- Ensembl REST — gene coordinates and metadata for ENSG00000012048
- gnomAD v4 — 7,279 variants with allele frequencies across the BRCA1 locus
- ClinVar — 500 records (retmax = 500) with clinical significance classifications
Variant Classification
ClinVar records were classified into "pathogenic" (including "pathogenic" and "likely pathogenic") and "benign" (including "benign" and "likely benign") categories. Records with conflicting classifications or ambiguous labels were excluded. This yielded 275 classified variants from the 500 fetched. Of these, 163 had both a genomic position suitable for joining with gnomAD and a non-zero gnomAD allele frequency; the remaining 112 were excluded either because they lacked genomic coordinates in ClinVar or because their gnomAD allele frequency was zero (not observed in the gnomAD sample). After joining, 39 were classified as pathogenic and 124 as benign.
Join Strategy
Variants were joined between ClinVar and gnomAD using genomic locus (chrom:pos), as ClinVar records frequently lack dbSNP rsIDs and ref/alt allele information. gnomAD variants at each locus were collapsed to the maximum allele frequency (representing the most common alt allele at multiallelic sites). This locus-level join is necessarily ref/alt-agnostic and may pair a ClinVar pathogenic allele with a different alt allele's frequency in gnomAD — a limitation we discuss below.
Statistical Test
A two-sided Mann-Whitney U test was applied to the allele frequencies of the pathogenic and benign groups. This non-parametric test was chosen because allele frequencies are heavily right-skewed. A fixed seed (1234) was used for all random operations (strip-plot jitter only; no randomness enters the statistic).
Results
After joining ClinVar and gnomAD by genomic locus, 163 variants had both a clinical classification and a gnomAD allele frequency > 0. Of these, 39 were classified as pathogenic and 124 as benign.
The Mann-Whitney U test did not detect a significant difference in allele frequencies between the two groups (U = 2621.5, p = 0.406). The median gnomAD allele frequency for pathogenic variants was 1.37 × 10⁻⁶, while the median for benign variants was 6.90 × 10⁻⁷. The observed direction (pathogenic variants having a numerically higher median frequency) is opposite to the expected pattern.
Figure: Distribution of gnomAD allele frequencies for ClinVar pathogenic (red, n = 39) and benign (blue, n = 124) BRCA1 variants. Box plots show medians and interquartile ranges; individual points are jittered variants.
Discussion
The null result — no detectable difference between pathogenic and benign variant frequencies in BRCA1 — contrasts with the textbook expectation that pathogenic variants should be rarer. Several factors may explain this.
First, both groups are extremely rare. BRCA1 is under strong purifying selection, and gnomAD allele frequencies for all classified variants are in the 10⁻⁶ to 10⁻⁵ range, near the detection limit of the current gnomAD sample size (~76,000 exomes/genomes). The distributions overlap substantially, reducing the power of any rank-based test.
Second, the locus-level join (chrom:pos without ref/alt) may mispair alleles. At a given locus, ClinVar may annotate one alternate allele as pathogenic while gnomAD reports a different alternate allele's frequency. This noise would attenuate any true signal, pushing toward the null.
Third, the ClinVar retmax cap of 500 means not all BRCA1 ClinVar records were retrieved, potentially biasing toward more frequently submitted (and possibly more common) pathogenic variants.
Fourth, the unexpected direction (pathogenic median AF numerically higher than benign) could reflect sampling variability given the small sample sizes (n = 39 and n = 124), or it could indicate that benign variants in BRCA1 are maintained at similarly rare frequencies because most of the coding space in this gene is functionally constrained.
Limitations
- The ClinVar × gnomAD join is at locus (chrom:pos) granularity without ref/alt matching, which may cause allele mispairing.
- The retmax cap of 500 ClinVar records means the full ClinVar BRCA1 corpus was not sampled.
- Summary-level data were used; individual-level genotypes were not accessed.
- Both groups are near the frequency detection floor of gnomAD, limiting statistical power.
- The analysis includes all variant types (missense, frameshift, synonymous, etc.) classified as pathogenic or benign; restricting to missense variants only might yield different results.
- Ensembl and gnomAD gene boundaries differ by 3 bp at the start coordinate (43044292 vs 43044295); this does not materially affect the analysis but is noted for completeness.
Provenance
| Dataset | Source | Accession | Access URL | Region Slice |
|---|---|---|---|---|
| Ensembl gene | Ensembl REST | ENSG00000012048 | rest.ensembl.org | 17:43044292–43170245 |
| gnomAD variants | gnomAD v4 | ENSG00000012048 | gnomad.broadinstitute.org/api | 17:43044295–43170245 |
| ClinVar records | NCBI ClinVar | BRCA1[gene] | eutils.ncbi.nlm.nih.gov | BRCA1 gene, 500 records |
Analysis: clinvar_gnomad_ensembl reference template, seed = 1234, Mann-Whitney U (two-sided). Script: anchor_clinvar_gnomad_ensembl.py.
clinvar gnomad ensembl
Acquisition → Analysis
-
Step 1 Ensembl fetcher
argvlookup--symbolBRCA1--out{{workspace:gene.json}}
declared outputsgene.json
-
Step 2 gnomAD fetcher
argv--geneBRCA1--datasetgnomad_r4--out{{workspace:gnomad.csv}}
declared outputsgnomad.csv
-
Step 3 ClinVar fetcher
argv--geneBRCA1--out{{workspace:clinvar.csv}}--retmax500
declared outputsclinvar.csv
Analysis
--clinvar{{workspace:clinvar.csv}}--gnomad{{workspace:gnomad.csv}}--geneBRCA1--seed{{seed}}--outdir{{workdir}}
cec97b1265e60cc06d59609e4cd3039f68c301a2f4589322f3acf46cbd169a9ffetched 2026-07-18T18:24:50Z · ClinVar (NCBI E-utilities)
089adee33f00ce3de70c04da90cf4656c158da91de958ed5f3356f7dc2fc3037fetched 2026-07-18T18:24:06Z · Ensembl REST
7596f3afb75920ab815732dde0959ca4bd697cabf7b7f2d113503ea3d65afa8dfetched 2026-07-18T18:24:27Z · gnomAD (gnomad_r4)
914cc037f6e7eb34be2ddfc77474c3d65b6e26711c0097c3bd76af912506b479
5625dd5f30547fb07997efeb3ab46d953d71f60c
{"gene":"BRCA1","seed":1234,"reference_template":"clinvar_gnomad_ensembl","join_key":"genomic locus chrom:pos (ref/alt-agnostic)","n_clinvar_classified":275,"n_merged_with_gnomad_af":163,"n_pathogenic":39,"n_benign":124,"outcome":"null_result","test":"Mann-Whitney U (two-sided) on allele frequency","u_statistic":2621.5,"p_value":0.4062813737023052,"median_af_pathogenic":0.00000137,"median_af_benign":0.00000069,"direction":"pathogenic not rarer","significant_at_0.05":false,"headline_statistic":"p = 4.06e-01 (Mann-Whitney U)"}