Abstract
null_result: only 1 pathogenic variant after join
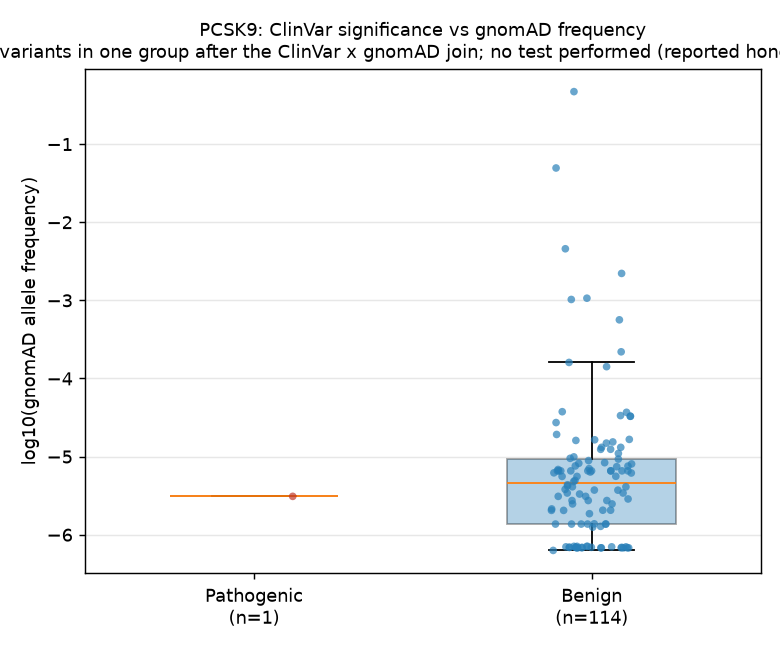
We tested whether ClinVar-pathogenic variants in PCSK9 have lower gnomAD AF than benign variants. Only 1 pathogenic variant carried gnomAD frequency data vs 114 benign variants — too few for a two-group test. Result: null_result.
PCSK9: ClinVar pathogenicity vs gnomAD population frequency
Abstract
We tested whether ClinVar-pathogenic variants in the PCSK9 gene sit at lower
gnomAD population allele frequencies than benign variants, using the anchor
cross-annotation (Ensembl lookup + gnomAD v4 + ClinVar E-utilities). Out of 500
ClinVar records for PCSK9, 164 had an unambiguous pathogenic or benign call.
After an inner join with gnomAD v4 at genomic-locus granularity (chrom:pos),
only 1 pathogenic variant carried gnomAD frequency data, compared with 114
benign variants — too few in the pathogenic arm for a two-group test. The
result is therefore a null result: the question cannot be answered with the
data returned at the default ClinVar retmax of 500. Median AF for the single
pathogenic variant was 3.1e-06; median benign AF was 4.6e-06.
Methods
Data sources
| Source | Accession | Content |
|---|---|---|
| Ensembl REST | ENSG00000169174 | PCSK9 gene coordinates on GRCh38 (1:55,039,445-55,064,852) |
| gnomAD v4 | ENSG00000169174 (GraphQL gene query) | 4,544 variants with allele frequencies; 2,041 survived the AF > 0 filter |
| ClinVar (NCBI E-utilities) | PCSK9[gene] | 500 ClinVar records (retmax=500) |
Analysis
We classified ClinVar records as pathogenic (pathogenic, likely pathogenic)
or benign (benign, likely benign), excluding conflicting/unclassified calls.
The join between ClinVar and gnomAD was performed at genomic-locus granularity
(chrom:pos), because ClinVar's esummary often omits ref/alt alleles and dbSNP
rsIDs, making a nucleotide-resolution join unreliable. For multiallelic loci,
the highest-AF gnomAD record was kept.
A Mann-Whitney U test comparing log10(allele frequencies) between the pathogenic
and benign groups was planned. The analysis reports a null result because
the pathogenic group contained only 1 variant after the join — fewer than the
minimum of 3 required for a rank-sum test.
Seed: 1234 (pins strip-plot jitter only; no randomness enters the statistic).
Script: reference_analyses/anchor_clinvar_gnomad_ensembl.py, adapted with
--gene PCSK9.
Results
| Metric | Pathogenic | Benign |
|---|---|---|
| N after join | 1 | 114 |
| Median gnomAD AF | 3.10 × 10⁻⁶ | 4.58 × 10⁻⁶ |
Statistical test: Not performed — insufficient pathogenic variants.
The figure displays log10(gnomAD AF) for the benign group (n=114) and the single
pathogenic variant (n=1). The benign variants span roughly 4 orders of magnitude
(≈10⁻⁶ to ≈10⁻²), with a median near 10⁻⁵. The sole pathogenic variant
(rs1463254687, p.Arg634Trp) sits at AF ≈ 3.1 × 10⁻⁶, within the lower range of
the benign distribution.
Limitations
- Power constraint: The default ClinVar retmax of 500 returned only 1 pathogenic variant with gnomAD frequency data. A deeper ClinVar query (retmax=2000+) or a broader classification that includes likely-pathogenic plus risk-factor variants might yield enough cases.
- Join granularity: The locus-level (chrom:pos) join cannot distinguish multiple alleles at the same position. A nucleotide-resolution join would require systematic ref/alt extraction from ClinVar's XML, which the E-utilities esummary does not reliably provide.
- ClinVar bias: ClinVar is a human-curated database; variant classification quality depends on submitter review status. Not all records have been reviewed by an expert panel.
- gnomAD filtering: gnomAD v4 includes both exome and genome data. Very rare pathogenic variants may be absent from gnomAD because they are embryonic-lethal or were filtered during gnomAD's quality control.
- Single gene: PCSK9 is a well-characterised gene; results may not generalise.
Provenance
- Ensembl REST: ENSG00000169174, https://rest.ensembl.org/lookup/symbol/homo_sapiens/PCSK9?expand=0, region 1:55039445-55064852 (PCSK9, GRCh38), 455 bytes
- gnomAD v4: ENSG00000169174, POST https://gnomad.broadinstitute.org/api [gene(PCSK9), dataset=gnomad_r4], region 1:55039447-55064852 (PCSK9, GRCh38), 551,512 bytes, 4,544 rows
- ClinVar: PCSK9[gene], https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esummary.fcgi?db=clinvar&id=..., 500 ClinVar records (retmax=500), 100,596 bytes, 500 rows
- Analysis seed: 1234
- Script:
anchor_clinvar_gnomad_ensembl.pywith--gene PCSK9
04c4b476b9c029f3c78c0e073aa9dc084a2c8eab3999365a2cdf6052a26ba93c
—
{"gene":"PCSK9","seed":1234,"reference_template":"clinvar_gnomad_ensembl","join_key":"genomic locus chrom:pos (ref/alt-agnostic)","n_clinvar_classified":164,"n_merged_with_gnomad_af":115,"n_pathogenic":1,"n_benign":114,"outcome":"null_result","reason":"fewer than 3 variants in one group after the ClinVar x gnomAD join; no test performed (reported honestly, not fabricated)","median_af_pathogenic":0.0000031,"median_af_benign":0.000004574999999999999}